Novel Mechanism for Oxygen Evolution Reaction on Dual-atom Catalysts Increases Reaction Activity
Atomically precise cluster catalysts bridge the gap between homogeneous and heterogeneous catalysis, facilitating comprehensive elucidation of structure-activity relationships in catalytic processes. However, the synergistic effects between non-single active sites in metal nanoclusters are scarcely explored.
In a study published in Nature Communications, a research team led by Prof. SUN Xiaoyan from the Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT) of the Chinese Academy of Sciences (CAS) provided pivotal mechanistic insights into the role of oxygen coupling between dual active sites during oxygen evolution reaction (OER).
In the conventional OER mechanism proceeded at a single active site, the electrochemical performance severely depends on the scaling relationship between the adsorption energies of oxygenated intermediates. Therefore, the bottleneck of the lowest theoretical overpotential of 0.37 V is difficult to overcome.
Inspired by the lattice oxygen mechanism, the research team systematically investigated the feasibility of *O-*O coupling mechanism (OCM) to improve the performance through high-throughput density functional theory (DFT) by using N-doped graphene supported dual-atom as a model catalyst, which can deviate from the scaling relationship of the conventional mechanism and consequently break through the bottleneck of 0.37 V.
“The findings not only provide fundamental insights into the non-single active site of cluster catalysts to enhance the reaction performance, but also help to provide theoretical guidelines for the rational design of other electrochemical reaction catalysts besides OER,” said Prof. SUN.
The research team further succeeded with efficiency and accuracy in predicting high-performance homonuclear dual-atom catalysts by means of rationally designed activity descriptors. The team also illustrated the conditions for the incidence of OCM as well as the OER performance of the constant potential method.
"This is the first time that high-throughput DFT has been employed to provide insight and understanding of unconventional OCM," said Prof. DING Yuxiao, another corresponding author to this study. "Not only that, a more sophisticated computational method enables predictions that are closer to real electrochemical conditions."
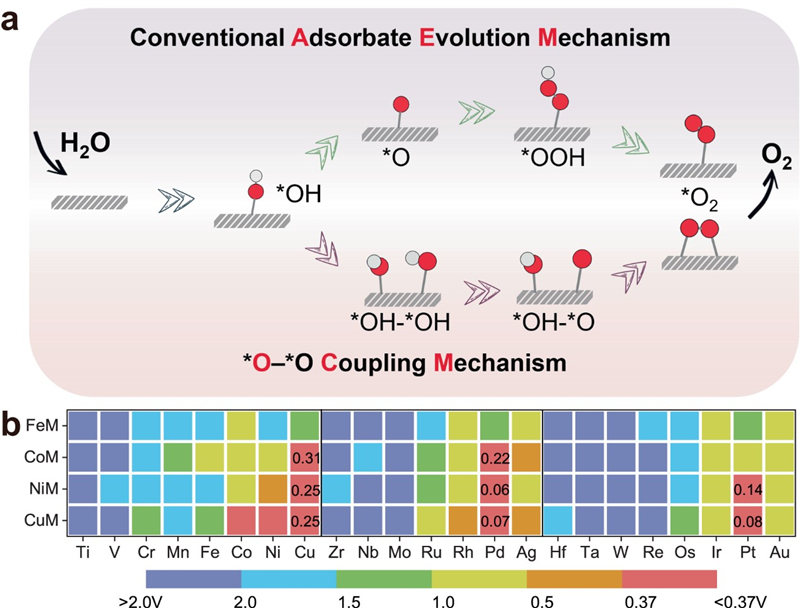
Schematic representation of the two possible mechanisms and reactivity map along the OCM. (Imaged by FANG Cong)
(Text by FANG Cong)
Contact:
KONG Fengru
Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences
Tel: 86-532-58261072
E-mail: kongfr@qibebt.ac.cn


