QIBEBT Unveiled Genome-wide Transcriptional Regulation Network of Oleaginous Microalgae Nannochloropsis
Microalgae are promising feedstock for fuels and chemicals because many species possess the ability to grow rapidly and synthesize large amounts of storage neutral lipids from sunlight and carbon dioxide. They can be cultivated on non-arable land with nonpotable water and waste streams and thus pose little competition to food crops while providing environmental benefits. However little was known about the transcriptional regulation networks in oleaginous microalgae, which has hindered the screening and engineering of microalgae for biofuels production.
In a study published in Scientific Reports on June 26, researchers from Single-Cell Center of Qingdao Institute of Bioenergy and Bioprocess Technology (QIBEBT), Chinese Academy of Sciences (CAS) unveiled the first genome-wide transcriptional regulation network for oleaginous microalgae and paved the way for genetic engineering for enhanced oil productivity in microalgae.
Nannochloropsis spp. is widely distributed in the marine environment as well as in fresh and brackish waters. It has been cultivated world-wide from small to large scale. The QIBEBT researchers has previously reported the genomes of six oleaginous Nannochloropsis strains (Wang, et al, PLoS Genetics, 2014) and a time-series transcriptomic dataset of oil accumulation process for N. oceanica strain IMET1 (Li, et al, Plant Cell, 2014).
Based on these experimental data and improvement on computational approaches such as Phylogenetic Footprinting, graduate student HU Jianqiang and his colleagues presented a genome-wide in silico map of transcription factors (TFs) and TFs binding sites (TFBSs). They also setup a computationally predicted and preliminary regulatory network that links TFs and target genes in Nannochloropsis. The network consists of 34 TFs, 30 TFBSs and 950 target genes (Figure 1A). Moreover, the group revealed transcriptional regulation of triacylglycerol (TAG) biosynthesis pathways that 11 TFs potentially involved in (Figure 1B). To allow readily access by the global research community, these findings are presented in an interactive online database (see Reference). Furthermore, the degree of similarity in TF-family profiles was found to be indicative of the phylogenetic relationship among the 36 surveyed plant and algal species. This suggested that the co-evolution of species and their TF-family profiles occurred largely at the level of TF-family.
This regulatory network forms a basis for genetic engineering of specific TFs and their target genes for enhanced microalgal biofuel production. It is also valuable to studies on the evolution of regulatory networks in plants.
The study was funded by a Synthetic Biology project from the National Basic Research Program of China (973 Program), and was jointly led by Professors XU Jian and NING Kang from the Single-Cell Center, QIBEBT, CAS.
Reference:
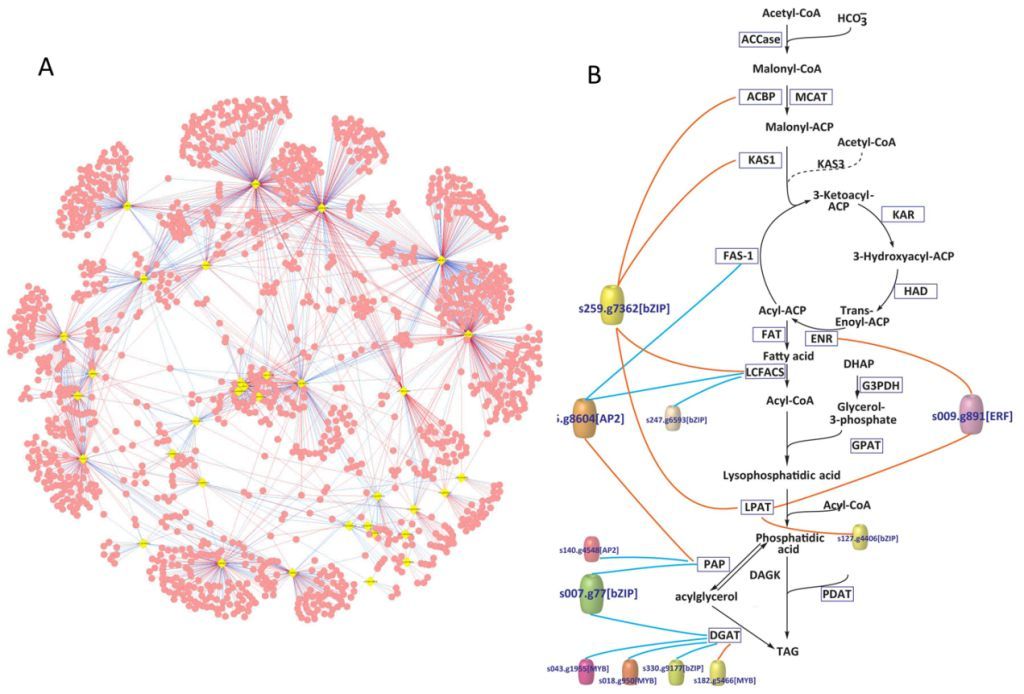
Figure 1: (A) An initial regulatory network of N.oceanica IMET1 based on the predicated TFs and TFBSs. Each “yellow quadrangle” represents a TF gene and each “red dot” represents a target gene. (B) The proposed transcriptional regulatory network of TAG biosynthesis in N. oceanica IMET1. Each “color cylinder” represents a TF gene. Red lines represent “positive” regulation and blue lines represent “negative” regulation. (Image by Single-Cell Center, QIBEBT)
1. Jianqiang Hu, Dongmei Wang, Jing Li, Gongchao Jing, Kang Ning, Jian Xu (2014) “Genome-wide identification of transcription factors and transcription-factor binding sites in oleaginous microalgae Nannochloropsis” Scientific Reports, 4 : 5454 | DOI: 10.1038/srep05454; http://www.nature.com/srep/2014/140626/srep05454/full/srep05454.html
2. The online database: http://www.singlecellcenter.org/en/NannoRegulationDatabase/home.htm
Contact:
Prof. XU Jian
Email: xujian (AT) qibebt.ac.cn
Web: http://english.qibebt.cas.cn


