High Performance Computing Applied to QIBEBT’s Computational Metagenomic Research
The microbial communities have played key roles in many bioenergy generation processes in nature, such as cellulose degradation and methane production. Metagenomics is based on direct sequencing of all microbes in the community, thus overcomes the difficulties in cultivation of microbes. As such, metagenomics has become one key approach for analyzing the structure and function of microbial communities. However, the large volume and complexity of metagenomic data has become one of the most critical bottlenecks.
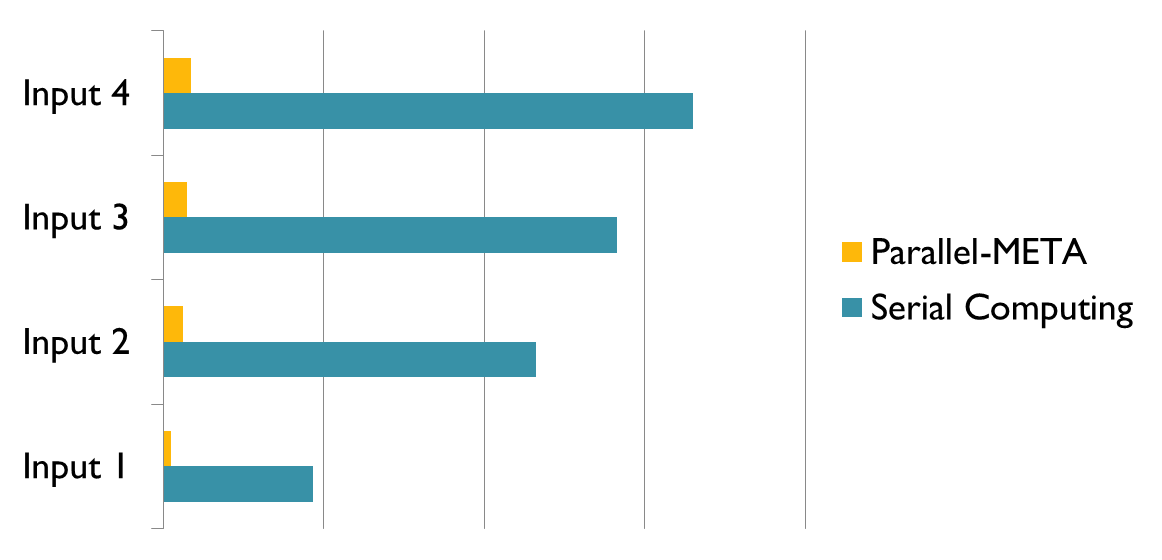 |
| Figure 1. Parallel-META is more than 10 times faster than traditional methods in analyzing the taxonomical structure of microbial communities (Image by SU Xiaoquan) |
Based on this fast and accurate taxonomical analysis method, the research team selected a broad ranges of metagenomic samples and datasets, and created a database (http://www.computationalbioenergy.org/meta-mesh.html). Based on this database, a series of methods for accurate comparison and search of metagenome samples were developed. These methods are useful for the identification of specific taxa and/or function from large number of metagenome samples. The papers were published in “Bioinformatics, 2012, 28 (19), 2494-2501” and “PLoS ONE, 2012, 7(11), e48998”.
In addition, the Functional Genomic Group has collaborated with Prof. CUI Xinping from University of California at Riverside, and Prof. ZHU Jiankang from Purdue University (Fellow of National Academy of Sciences, USA) to develop a novel method for single nucleotide polymorphism (SNP) detection called GeMS (http://www.computationalbioenergy.org/snp.html ). This method should find broad applications in evolution, mutation analysis, etc.
The Metagenomics Platform (http://www.bioenergychina.org/fg/ , http://www.computationalbioenergy.org ) at Functional Genomic Group, which is based on the above mentioned methods and database, has been supporting collaborative microbiome research from over 30 institutes, universities and corporations. Through this collaborative network, these new computational methods are supporting diverse application areas such as bioenergy, environment, healthcare, agriculture, etc.
The research works were supported by MoST of China (863 project) and National Scientific Foundation of China.
References:
1. PLoS ONE, 2012, 7(11), e48998
2. Bioinformatics, 2012, 28 (19), 2494-2501,
3. BMC Systems Biology, 2012, 6 (Suppl 1): S16
Contact:
Dr. NING Kang, Associate Professor
Qingdao Institute of Bioenergy and Bioprocess Technology
Chinese Academy of Sciences
Tel: +86-(0)532-80662624
E-mail: ningkang@qibebt.ac.cn
Website: http://www.bioenergychina.org/fg/index.html


